

CMT is a genetically heterogenous disorder which effects the motor and sensory peripheral nerves and may also be known clinically as Hereditary Motor and Sensory Neuropathy (HMSN); in the rare occasion where it only effects motor nerves, it is recognised as Distal Hereditary Motor Neuro(no)pathy (dHMN) (Pareyson & Marchesi, 2009). It is one of the most common hereditary neuromuscular disorders with an occurrence of 40 incidences in every 100,000 individuals (Martyn & Hughes, 1997). CMT is caused by mutation of genes responsible for encoding various proteins such as compact/non-compact myelin, Schwann cells and axons which are involved in everyday functions of the body such as mitochondrial metabolism, axonal transport and maintenance of myelin (Barisic et al., 2008; Szigeti & Lupski, 2009; Hoogendijk et al., 1994). Regardless of the defects that primarily effect the myelin or axon, the common end pathway seen of CMT is degenerative process at the axon involving the largest and longest fibres (Pareyson, Scaori & Laura, 2006; Schrerer & Wrabetz, 2009; Krajewski et al., 2000).
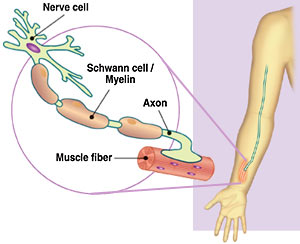
Figure 1. Visual representation of the location of the axon and myelin (nawrot.psych.ndsu.nodak.edu)
Unlike early onset foot conditions such as Congenital Talipes Equinovarus (Clubfoot) which is treated within the first years of birth (Dobbs & Gurnett, 2009), CMT usually occurs over the first 2 decades of life and then progresses relatively thereafter over decades, and neither is it restricted to the feet as it may progress from the feet to the entire lower limb region and even the upper limbs in some subtypes of the condition (Pareyson & Marchesi, 2009).
Though the diagnostic process may be complicated, there are at least 25 specific genes that have been identified to be associated with CMT and 70% of patients are now able to receive a precise molecular genetic diagnosis. The process generally includes identification and definition of the clinical phenotype, identification of the inheritance pattern, electrophysiological examination and a molecular analysis (Pareyson & Marchesi, 2009). The molecular diagnosis can be significantly aided by clinical information/findings of the patients including the age of CMT onset, severity of the disease and presence of uncommon features which still may be associated with CMT such as optic atrophy, vocal cord palsy and glaucoma (Shy et al, 2005; Pareyson, Scaori & Laura, 2006; Barisic et al., 2008; Reilly, 2007; Szigeti & Lupski, 2009).
Based on nerve conduction studies and nerve pathology, CMT can be classified into two types: Demyelinating CMT which is characterised by slow nerve conduction velocities (<35 m/s in the upper limb motor nerves) and is further split into CMT-1 or CMT-4. This split is based on inheritance identification, with CMT-1 presenting itself as autosomal dominant (defect in a dominant non sex specific chromosome) and CMT-4 as autosomal recessive (defect in a recessive non sex specific chromosome). The second type of CMT is axonal and these are characterised by relatively faster nerve conduction velocities (>35 m/s) and pathological evidence of chronic axonal degeneration. Autosomal dominant forms such as CMT-1, CMT-2, dHMN are the most common expressions of CMT in cases while autosomal recessive forms are slightly more rare and severe with an early onset and can be either demyelinating or axonal such as CMT-4, AR-CMT-2 and AR-dHMN. (Ouvrier, Geevasingha, & Ryan, 2007; Vallat, Tazir, Magdelaine, & Sturtz, 2005). However, with the knowledge of CMT being consistently further enhanced, research has begun to find many subtypes in between some of the above mentioned, most importantly CMT-X or x linked CMT (CMT-X1), which is a form of the disease carried by the parent in the x-chromosome (responsible for determining the child’s sex) and is then passed down. This type of CMT cannot be transferred from male to male (therefore a son cannot inherit this from his father) but it is still found to be more common in men (Pareyson, Scaori & Laura, 2006; Barisic et al., 2008).

Figure 2. Some of the most common symptoms of CMT
Due to most forms of CMT being an autosomal disorder, it is not specific to gender and effects both men and women equally (Dyck, Chance, Lebo & Carney, 1993). All muscles and muscle fiber types are effected with the distal muscles being effected the most severely (Tsairis, 1974; Borg & Ericson-Gripenstedt, 2002; Erikson, Ansved & Borg, 1998). Symptoms include muscle wastage, muscle weakness, atrophy in the lower limb muscles (particularly the calves), reduced (or absent) deep tendon reflexes, change in gait with difficulty in walking/running, arched feet with development of ‘hammer toes’ and muscle cramps. In some cases, the hands are also affected, along with the fore arms, leading to hand tremors and a clawed hand posture. Sensory loss generally follows a similar pathway (from lower to upper limb) causing loss of sensation within the feet and hands in terms of pain, vibration and touch (Shy et al, 2005; Pareyson, Scaori & Laura, 2006; Barisic et al., 2008; Jani-Acsadi, Krajewski, & Shy, 2008; Reilly, 2007; Szigeti & Lupski, 2009). Onset has been recorded in some cases at such early age that it has caused hypotonia (‘floppy baby syndrome’) and delayed motor development, whilst in other cases, CMT has not fully expressed itself until much later on in life (Pareyson & Marchesi, 2009).
Rehabilitative Considerations
Unfortunately, there is currently still no effective or known drug therapy for CMT, leaving treatment limited to rehabilitation therapy or surgical procedures in the case of skeletal deformities or soft tissue abnormalities, while clinical and animal trials are still currently in the works (Young, De Jonghe, Stögbauer, & Butterfass‐Bahloul, 2008; Sackley et al, 2009). This means the management of CMT in patients requires an interdisciplinary approach with the collaboration of a neurologist, physiotherapist/qualified strength coach and other professionals (Erikson, Ansved & Borg, 1998).
The biggest implication of training patients with a neuromuscular disease such as CMT is the ‘overwork hypothesis’ within which research has pointed towards weakness being further induced from resistance training or general overload (Vinci et al., 2003; van Pomeren et al., 2009). This originated from the study of Kilmer et al., (1994) who observed an increase in injury after a high intensity home based resistance training programme, and concluded that increases in training frequency, volume and particularly intensity is a major risk for patients suffering a neuromuscular disease. Lindeman et al., (1995) found improvements in strength and functional ability after a moderate -high intensity training programme in CMT patients, suggesting that high intensity training is possibly a grey area for training prescription.
Chetlin et al., (2004) observed a twelve week home based resistance training programme focused on improving strength, body composition and activities of daily living, and found that activities of daily living and strength were significantly improved from baseline in both men and women. The programme used in the experimental design placed emphasis on the knee and elbow extensors and flexors, with resistance exercises such as tricep extensions, bicep curls, leg extensions and leg curls. This suggests that moderate exercises can be safe and effective for CMT patients to improve strength and performance in day to day activities.
Though it is yet to be a proven method of rehabilitative therapy for CMT patients, it has been theorised that passive stretching of the ankle flexors and extensors could be prescribed within programming to help counteract tendon retractions and improve it’s reflex ability (Refshauge et al., 2006). Matjacić and Zupan, (2006) also observed the effects of passive stretching along with exercises aimed at general muscle strengthening and balance either guided by a physical therapist or by the set balance apparatus, over a twelve-session intervention and concluded postural and dynamic balance training to be a useful training modality to improve balance and mobility. They suggested this may be due to an improvement in utilisation of compensatory balance and movement strategies of the proximal muscle groups as the distal lower limbs wiuld have been significantly weakened due to CMT.
Patients with CMT have also been reported to present reduced peak oxygen consumption (Vo2 Max) values and a generally decreased aerobic capacity and it has been suggested moderate aerobic exercise may improve functional ability and aerobic capacity but this is yet to be further studied and proven (El Mhandi et al., 2007).
In summary, though it may be difficult for a strength coach, physiotherapist or patient to conclude much on exercise prescriptions and recommendations from this brief overview on the current literature in the field of CMT, it is however safe to conclude:
- Resistance exercise can be beneficial if the patients level of weakness is not severe, and if the rate of progression of the disease is relatively slow.
- High-intensity resistance exercises/programmes have no advantage over moderate intensity training programmes (Kilmer, 2002).
References:
Barisic, N., Claeys, K. G., Sirotković‐Skerlev, M., Löfgren, A., Nelis, E., De Jonghe, P., & Timmerman, V. (2008). Charcot‐Marie‐Tooth Disease: A Clinico‐genetic Confrontation. Annals of human genetics, 72(3), 416-441.
Borg, K., & Ericson-Gripenstedt, U. (2002). Muscle biopsy abnormalities differ between Charcot-Marie-Tooth type 1 and 2: reflect different pathophysiology?. Exercise and sport sciences reviews, 30(1), 4-7.
Chetlin, R. D., Gutmann, L., Tarnopolsky, M. A., Ullrich, I. H., & Yeater, R. A. (2004). Resistance training exercise and creatine in patients with Charcot–Marie–Tooth disease. Muscle & nerve, 30(1), 69-76.
Dyck, P., Chance, P., Lebo, R., Carney, J. (1993). Hereditary motor and sensory neuropathies. In: Dyck P, Thomas P, editors. Peripheral neuropathy, 3rd ed. Philadelphia: WB Saunders; p 1094- 136.
Dyck, P. J., Karnes, J. L., & Lambert, E. H. (1989). Longitudinal study of neuropathic deficits and nerve conduction abnormalities in hereditary motor and sensory neuropathy type 1. Neurology, 39(10), 1302-1302.
El Mhandi, L., Calmels, P., Camdessanché, J. P., Gautheron, V., & Féasson, L. (2007). Muscle strength recovery in treated Guillain-Barré syndrome: a prospective study for the first 18 months after onset. American Journal of Physical Medicine & Rehabilitation, 86(9), 716-724.
Ericson, U., Ansved, T., & Borg, K. (1998). Charcot‐Marie‐Tooth disease type 1 and 2‐an immunohistochemical study of muscle fibre cytoskeletal proteins and a maker for muscle fibre cytoskeletal proteins and a marker for muscle fibre regeneration. European journal of neurology, 5(6), 545-551.
Herrmann, D. N. (2008). Experimental therapeutics in hereditary neuropathies: the past, the present, and the future. Neurotherapeutics, 5(4), 507-515.
Hoogendijk, J. E., de Visser, M., Bolhuis, P. A., Hart, A. A., & de Visser, B. W. O. (1994). Hereditary motor and sensory neuropathy type I: clinical and neurographical features of the 17p duplication subtype. Muscle & nerve, 17(1), 85-90.
Houlden, H., Laura, M., Wavrant–De Vrièze, F., Blake, J., Wood, N., & Reilly, M. M. (2008). Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology, 71(21), 1660-1668.
Jani-Acsadi, A., Krajewski, K., & Shy, M. E. (2008, April). Charcot-Marie-Tooth neuropathies: diagnosis and management. In Seminars in neurology (Vol. 28, No. 02, pp. 185-194). © Thieme Medical Publishers.
Kilmer, D. D. (2002). Response to resistive strengthening exercise training in humans with neuromuscular disease. American journal of physical medicine & rehabilitation, 81(11), S121-S126.
Kilmer, D. D., Wright, N. C., & Aitkens, S. (2005). Impact of a home-based activity and dietary intervention in people with slowly progressive neuromuscular diseases. Archives of physical medicine and rehabilitation, 86(11), 2150-2156.
Kilmer, D. D., McCrory, M. A., Wright, N. C., Aitkens, S. G., & Bernauer, E. M. (1994). The effect of a high resistance exercise program in slowly progressive neuromuscular disease. Archives of physical medicine and rehabilitation, 75(5), 560-563.
Krajewski, K. M., Lewis, R. A., Fuerst, D. R., Turansky, C., Hinderer, S. R., Garbern, J., & Shy, M. E. (2000). Neurological dysfunction and axonal degeneration in Charcot–Marie–Tooth disease type 1A. Brain, 123(7), 1516-1527.
Lindeman, E., Leffers, P., Spaans, F., Drukker, J., Reulen, J., Kerckhoffs, M., & Köke, A. (1995). Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Archives of physical medicine and rehabilitation, 76(7), 612-620.
Martyn, C.N., Hughes, R.A.C. (1997). Epidemiology of peripheral neuropathy. Journal of Neurology, Neurosurgery and Psychiatry. 62: 310–318.
Matjacić, Z., & Zupan, A. (2006). Effects of dynamic balance training during standing and stepping in patients with hereditary sensory motor neuropathy. Disability and rehabilitation, 28(23), 1455-1459.
Ouvrier, R., Geevasingha, N., & Ryan, M. M. (2007). Autosomal‐recessive and X‐linked forms of hereditary motor and sensory neuropathy in childhood. Muscle & nerve, 36(2), 131-143.
Pareyson, D., Scaioli, V., & Laurà, M. (2006). Clinical and Electrophysiological Aspects of Charcot-Marie-Tooth Disease. NeuroMolecular Medicine,8(1-2), 3-22.
Raeymaekers, P., Timmerman, V., Nelis, E., De Jonghe, P., Hoogenduk, J. E., Baas, F., & Van Broeckhoven, C. (1991). Duplication in chromosome 17p11. 2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscular Disorders, 1(2), 93-97.
Refshauge, K. M., Raymond, J., Nicholson, G., & van den Dolder, P. A. (2006). Night splinting does not increase ankle range of motion in people with Charcot-Marie-Tooth disease: a randomised, cross-over trial. Australian Journal of Physiotherapy, 52(3), 193-199.
Reilly, M. M. (2007). Sorting out the inherited neuropathies. Practical neurology, 7(2), 93-105.
Sackley, C., Disler, P. B., Turner‐Stokes, L., Wade, D. T., Brittle, N., & Hoppitt, T. (2009). Rehabilitation interventions for foot drop in neuromuscular disease. The Cochrane Library.
Scherer, S. S., & Wrabetz, L. (2008). Molecular mechanisms of inherited demyelinating neuropathies. Glia, 56(14), 1578-1589.
Shy, M. E. (2006). Therapeutic strategies for the inherited neuropathies. Neuromolecular medicine, 8(1-2), 255-278.
Shy, M., Lupski, J.R., Chance, P.F., Klein, C.J., Dyck, P.J. (2005). Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic and pathologic features. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy 4th edn. Philadelphia: Elsevier Saunders. 1623–58.
Szigeti, K., & Lupski, J. R. (2009). Charcot–Marie–Tooth disease. European Journal of Human Genetics, 17(6), 703-710.
Szigeti, K., Garcia, C. A., & Lupski, J. R. (2006). Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genetics in Medicine, 8(2), 86-92.
Tsairis, P. (1974). Muscle Biopsy: A Modern Approach. Archives of Neurology, 31(2), 143-143.
Vallat, J. M., Tazir, M., Magdelaine, C., & Sturtz, F. (2005). Autosomal-recessive Charcot-Marie-Tooth diseases. Journal of Neuropathology & Experimental Neurology, 64(5), 363-370.
van Pomeren, M., Selles, R. W., van Ginneken, B. T., Schreuders, T. A., Janssen, W. G., & Stam, H. J. (2009). The hypothesis of overwork weakness in Charcot-Marie-Tooth: a critical evaluation. Journal of rehabilitation medicine, 41(1), 32-34.
Vinci, P., Esposito, C., Perelli, S. L., Antenor, J. A. V., & Thomas, F. P. (2003). Overwork weakness in Charcot-Marie-Tooth disease. Archives of physical medicine and rehabilitation, 84(6), 825-827.
Young, P., De Jonghe, P., Stögbauer, F., & Butterfass‐Bahloul, T. (2008). Treatment for Charcot‐Marie‐Tooth disease. The Cochrane Library.
